Clinical Definition
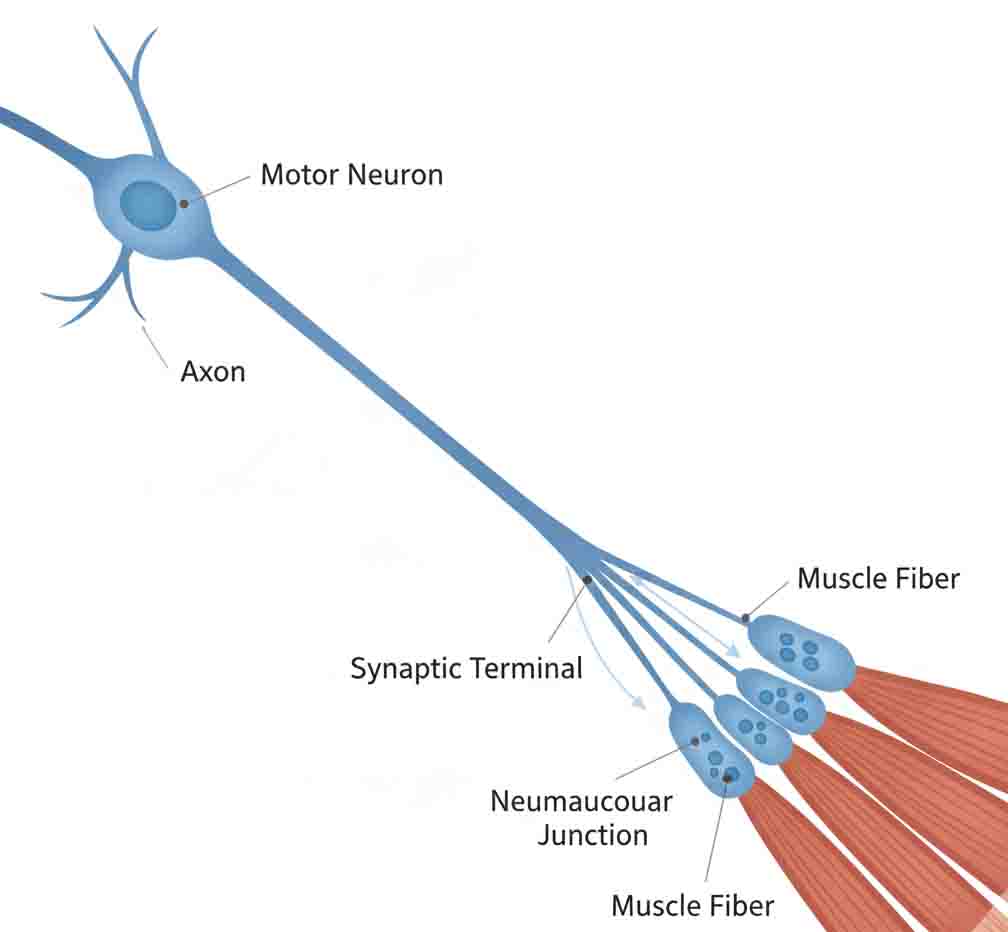
Amyotrophic Lateral Sclerosis (ALS), also termed Motor Neuron Disease (MND), is a progressive neurodegenerative disorder characterized by the selective apoptosis of both Upper Motor Neurons (UMN) in the motor cortex and Lower Motor Neurons (LMN) in the brainstem and spinal cord. This dual degeneration leads to progressive muscle atrophy, fasciculations, spasticity, and eventually respiratory failure. Etiologically, it is classified into Sporadic ALS (SALS, 90-95%) and Familial ALS (FALS, 5-10%).
Clinical Coding & Classification
| System / Category | Code(s) | Description |
|---|---|---|
| ICD-10-CM | G12.21 | Amyotrophic lateral sclerosis |
| ICD-10-CM | G12.2 | Motor neuron disease (Category) |
| CPT (Diagnostic) | 95860 – 95872 | Needle electromyography (EMG) |
| CPT (Diagnostic) | 95907 – 95913 | Nerve conduction studies (NCS) |
| Affected System | Neurological | Corticospinal Tract; Anterior Horn Cells |
Epidemiology & Statistics
ALS is the most common adult-onset motor neuron disease, with an incidence of approximately 1.7 to 2.3 per 100,000 person-years and a prevalence of 5 per 100,000. The mean age of onset for sporadic cases is 58-63 years, while familial cases present earlier. There is a slight male predominance (1.5:1 ratio). Life expectancy averages 2-5 years from diagnosis, primarily due to respiratory muscle paralysis.
Pathophysiology (Mechanism)
The pathogenesis is multifactorial, involving complex molecular cascades:
1. Glutamate Excitotoxicity: Impaired clearance of glutamate by astrocytes (EAAT2 transporter defect) leads to calcium influx and neuronal death.
2. Protein Aggregation: Accumulation of misfolded proteins, most notably TDP-43 (seen in 97% of cases) and SOD1 (Superoxide Dismutase 1) mutations, causing intracellular inclusions.
3. Oxidative Stress: Mitochondrial dysfunction leads to the accumulation of reactive oxygen species (ROS), damaging cellular lipids and DNA.
4. Genetic Drivers: Key mutations include C9orf72 (most common genetic cause) and SOD1.
Standard Management Protocols
Management is palliative and stage-dependent, focusing on slowing progression and symptom control.
- Pharmacological Classes (Disease-Modifying):
- Riluzole: Glutamate release inhibitor. Proven to extend survival by 2-3 months. Standard of care.
- Edaravone: Free radical scavenger. Shown to slow functional decline in specific patient subsets.
- Antisense Oligonucleotides (ASOs): (e.g., Tofersen) Emerging gene therapy targeting SOD1 mutations.
- Supportive Interventions:
- Non-Invasive Ventilation (NIV): (e.g., BiPAP) Critical for managing respiratory insufficiency and prolonging survival.
- Gastrostomy (PEG Tube): Indicated for severe dysphagia to maintain caloric intake.
Healthcare Resource Utilization
ALS care is resource-intensive due to the need for multidisciplinary management:
- Durable Medical Equipment (DME): High utilization of power wheelchairs, speech-generating devices (eye-tracking technology), and home ventilators.
- Home Health Care: Increasing dependency requires significant nursing and caregiver support as the disease progresses to tetraplegia.